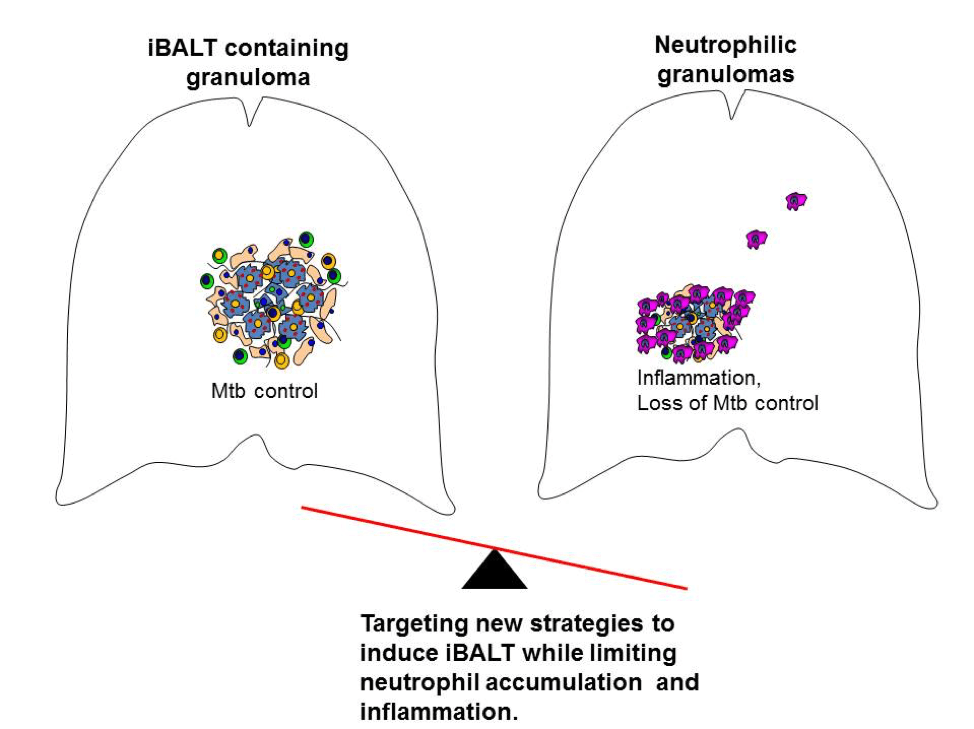
Mycobacterium tuberculosis (Mtb), the causative agent of the disease tuberculosis (TB), is estimated to infect one-fourth of the world’s population, resulting in approximately 1.5 million deaths each year. The emergence of multidrug- and extensively drug-resistant Mtb strains and the variable efficacy of the currently used vaccine, M. bovis Bacille Calmette Guerin (BCG), are barriers to the global control of TB. Thus, there is a critical need to better understand the mechanisms of TB immunopathogenesis, as such mechanisms can be targeted to improve host control of Mtb infection. Following inhalation of aerosolized Mtb, bacteria are phagocytosed by alveolar macrophages (AMs) and dendritic cells (DCs), resulting in the recruitment of immune cells to the lung and the formation of the tubercle granuloma. Although granulomas have long been considered a hallmark of both latent TB infection (LTBI) and clinical pulmonary TB (PTB), the immunological differences between protective granulomas and non-protective granulomas have only recently begun to emerge. Our recent data suggest that the presence of B-cell follicles in inducible bronchus-associated lymphoid tissue (iBALT)-containing granulomas is indicative of protective granulomas that mediate Mtb control during LTBI. In contrast, infiltrating neutrophils producing proinflammatory molecules are characteristic of non-protective granulomas during PTB. Our findings challenge the current paradigm that TB granulomas are generally protective and advocate a new model for TB immunopathogenesis in which protective granulomas contain iBALT whereas non-protective granulomas are neutrophilic or exhibit immune activation. We will further elucidate the mechanisms by which iBALT-containing granulomas confer protection against TB and will test approaches for clinically improving iBALT formation and limiting neutrophil accumulation. The knowledge to be gained from these studies is significant, as it will advance the development of new therapeutics and vaccine strategies for limiting TB-Reactivation (TB-R) and progression to PTB.
The presence of B cell follicles in iBALT is associated with protection in TB, but the mechanism behind the functional role of B cells in TB is still lacking. Mice lacking B cells are more susceptible to Mtb infection, and show greater immunopathology, indicating that B cells have a protective role in Mtb infection. Our aim is to further elucidate and identify the roles B cells are playing in protection against Mtb infection. To accomplish our aim, we will use different Mtb-infected mouse strains lacking particular B cell subsets or functions, and characterize their response to Mtb infection. Ultimately, our objective is to understand the mechanisms through which B cells can protect against Mtb infection, and identify targets which we can utilize to combat TB.
The role of neutrophils, the predominant cells harboring replicating Mtb during TB, is being actively explored and its accumulation is associated with lung pathology. Using the well-established mouse model of TB, the role of constitutively expressed neutrophilic protein, S100A8/A9 mediated neutrophil accumulation during progression to chronic TB was established (Gopal et al. 2013). The abrogation of neutrophils or S100A8/A9 improves Mtb control during chronic phase of the disease. Extensive collaborative work conducted in the NHP model of TB with the Kaushal Lab, Texas Biomedical Research Institute, and in human studies has substantiated our findings in the mouse model. The focus of our ongoing studies is to exploit the signaling pathways that control S100A8/A9 expression and target it to decrease Mtb disease susceptibility to enable host directed therapy for TB. Additionally, S100A8/A9 in conjunction with other chemokine markers in blood serum / urine are being explored as candidate targets to develop a rapid diagnostic test for TB.
Our laboratory is interested to study the function of ILCs, which are the innate counterpart of T cells. ILCs are present at the mucosal site where they quickly initiate cytokine response against pathogens. In 2019, in a seminal study published in Nature, we along with the Leslie Lab, AHRI, showed that an IL-17/IL-22 producing ILC subset, namely ILC3s, play a crucial role in early innate immune response to TB. We identified CXCR5 mediated mechanism of ILC3 accumulation within the iBALT structure of TB granuloma (Ardain et al. 2019). We also investigated the role of transcription factor, aryl hydrocarbon receptor (ahr) in ILC3 development in collaboration with the laboratory of Dr. Marco Colonna. We are investigating the transcriptome and epigenome of ILCs modified during TBs (also in vaccine model). We are also interested in studying the ILC plasticity in the murine model of tuberculosis.
Our work focuses on the early innate immune response to PTB infection. More specifically, this project focuses on the early interactions between tissue resident AM and other cell types such as lung epithelial cells (EC), recruited myeloid and lymphoid cells. Furthermore we aim to determine how these cellular interactions and the physical localization of AMs determines their effector functions during pulmonary Mtb infection.
We developed a novel technique for labeling immune cells present in the airway or the vasculature using fluorophore conjugated antibodies administered by intratracheal (i.t.) instillation and intravenous injection (i.v.) prior to harvest, allowing us to determine not only the number and percentage of specific cell types by flow cytometry, but also their specific location in the lung at the time of harvest. Using this cutting edge technique, we demonstrated for the first time that during pulmonary Mtb infection with a hypervirulent clinical isolate of Mtb, AMs have the ability to egress from the airway to the interstitial and parenchymal lung tissue over time, guided by the CCR2-CCL2 axis, and that they have the ability to localize within TB granulomas in a CCR2 dependent manner (Dunlap et al. 2018). From there we found that airway and non-airway AMs have distinct transcriptional profiles marking different effector functions, where non-airway AMs have a marked increase in M1 profile associated genes involved in phagocytosis, complement interactions, iNOS mediated killing, MHCII driven antigen presentation and T cell interactions. These data demonstrated for the first time that tissue resident AMs have the ability to migrate during Mtb infection and have compartmentally distinct functional roles.
Vaccines delivered through the mucosal route are known to induce T helper type 17 (Th17) responses and provide superior protection against Mtb infection. However, already tested Th17-inducing mucosal adjuvants such as heat labile enterotoxins and cholera toxins, are not considered safe for use in humans. To address these issues we have rationally screened adjuvants and identified combined use of MPL and chitosan as a novel formulation for use as a mucosal adjuvant to generate protective immune responses against Mtb infection. The fact that this vaccine formulation is protective against emerging clinically relevant Mtb strains such as the hypervirulent Mtb HN878 strain, further supports the use of MPL-chitosan as a potent Th17 inducing adjuvant for mucosal use in human TB vaccines.
As the development of safe mucosal adjuvants for human use is critical, we have further demonstrated that nanoemulsion (NE)-based adjuvants when delivered intranasally along with Mtb specific immunodominant antigens (NE-TB vaccine) induce potent mucosal IL-17 T-cell responses. Additionally, the NE-TB vaccine confers significant protection against Mtb infection, and when delivered along with BCG, is associated with decreased disease severity (Ahmed et al. 2017). These findings strongly support the development of a NE-TB vaccine as a novel, safe and effective, first-of-kind IL-17 inducing mucosal vaccine for potential use in humans. The NE-TB technology has been submitted for a patent at the Office of Technology Management – Washington University in St. Louis. Further investigations and trials with primate and guinea-pig models are ongoing and will be very crucial toward achieving higher goals and translation of the NE-TB vaccine for use in a human TB vaccine.
The development of a TB vaccine that induces sterilizing immunity to Mtb infection has been elusive. Absence of sterilizing immunity induced by TB vaccines may be due to delayed activation of mucosal DCs, and subsequent delay in antigen presentation and activation of vaccine-induced CD4+T-cell responses. Our published studies have shown that pulmonary delivery of activated Mtb antigen-primed DCs into vaccinated mice, at the time of Mtb exposure, can overcome the delay in accumulation of vaccine-induced CD4+ T-cell responses. In addition, activating endogenous host CD103+ DCs and the CD40-CD40L pathway can similarly induce rapid accumulation of vaccine-induced lung CD4+T-cell responses and limit early Mtb growth (Griffiths et al. 2016). Thus, targeting mucosal DCs can accelerate vaccine induced T-cell responses on Mtb infection, and provide insights to overcome bottlenecks in TB vaccine efficacy.
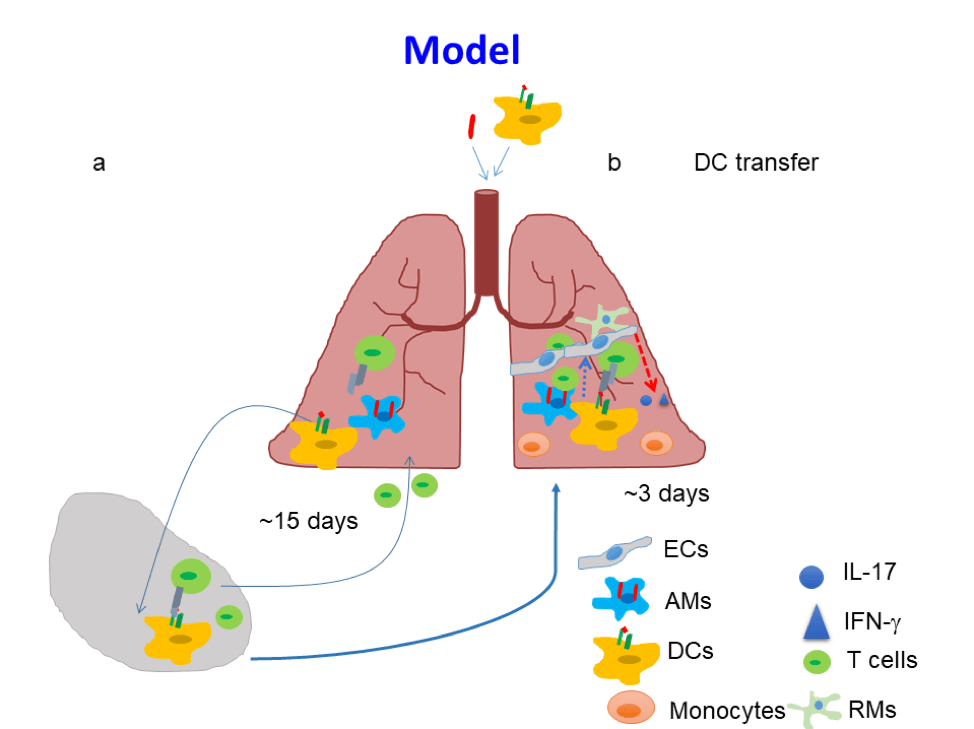
Induction of sterilizing immunity via pulmonary delivery of activated Mtb antigen-primed DCs
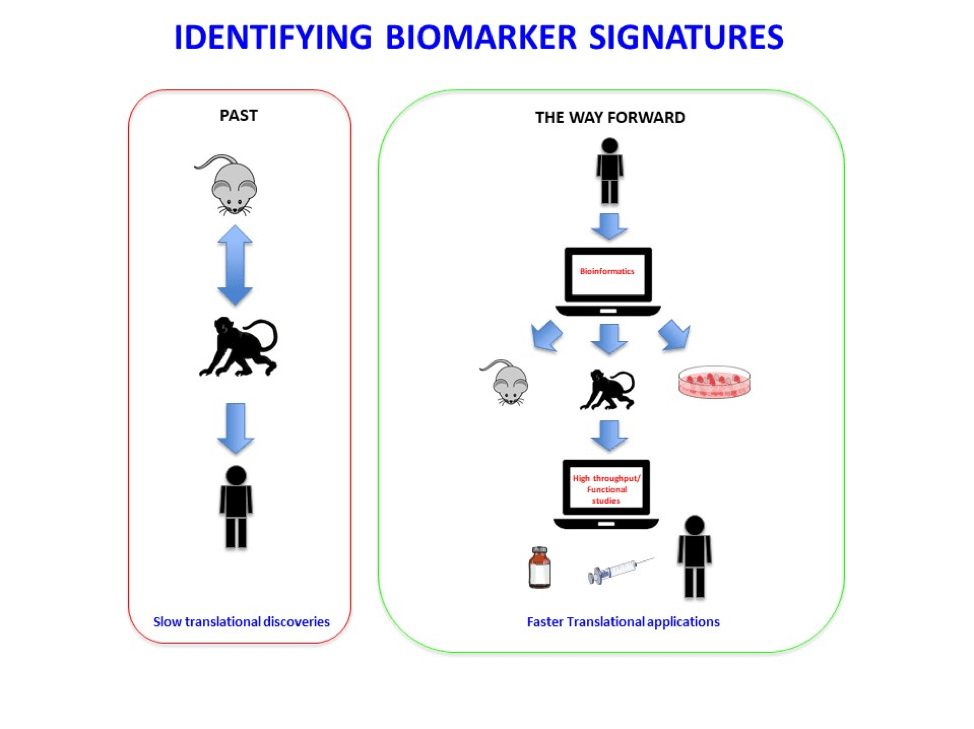
Animal models have been extensively used to characterize immune mechanisms of TB. However, immune correlates of protection or inflammation identified using animal models have not been validated in human studies. Immune correlates identified using animal models are not always effectively translated to human TB, thus resulting in a slow pace of translational discoveries from animal models to human TB for many platforms including vaccines, therapeutics, biomarkers and diagnostic discovery. Therefore, it is critical to improve our poor understanding of immune correlates of disease and protection that are shared across animal TB models and human TB. We have investigated the common immune correlates of risk of TB disease across animal models and humans and have mechanistically interrogated the functional role of these genes in Mtb infection using diversity outbred mouse model along with gene deficient mouse models. We have provided an in-depth, unbiased identification of the conserved and diversified gene/immune pathways in non-human primate and diversity outbred mouse TB models and human TB. The results from our studies show for the first time that in fact prominent differentially expressed genes/pathways induced during TB disease progression are conserved in genetically diverse mice, macaques and humans. Additionally, using gene deficient mouse models, we have addressed the functional role of individual genes comprising the gene signature of disease progression seen in humans with Mtb infection. Our data show that genes representing specific immune pathways can be protective, detrimental or redundant in controlling Mtb infection, and translate into identifying new immune pathways that mediate TB immunopathology in humans (Ahmed et al. 2020). Together, our cross species findings provide novel insights into modeling TB disease and the immunological basis of TB disease progression.
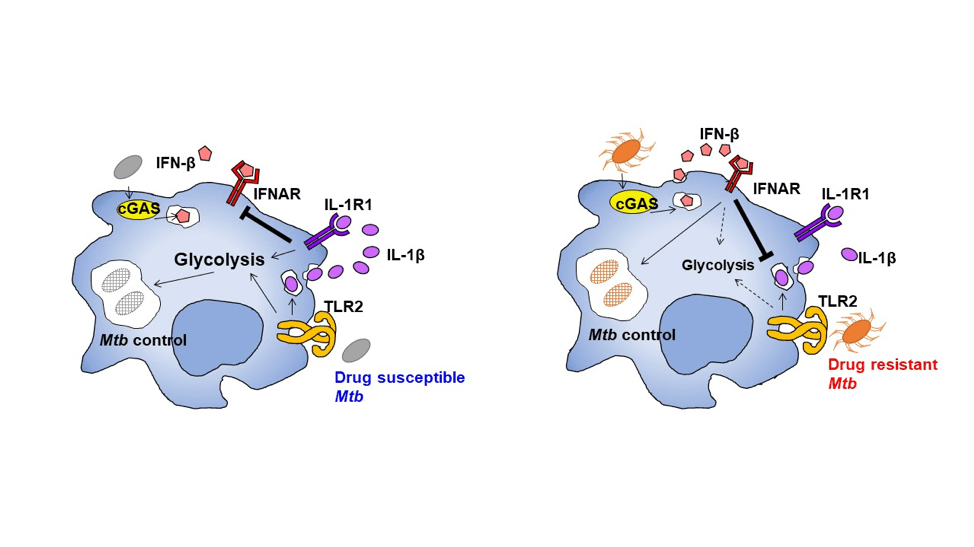
The emergence of multidrug-resistant (MDR) Mtb that is resistant to the frontline anti-tubercular drugs rifampicin and isoniazid forces treatment with toxic second-line drugs. Currently, ~4% of new and ~21% of previously treated tuberculosis cases are either rifampicin-drug-resistant or MDR Mtb infections. The specific molecular host-pathogen interactions mediating the rapid worldwide spread of MDR Mtb strains remain poorly understood. W-Beijing Mtb strains are highly prevalent throughout the world and associated with increased drug resistance. The production of interleukin-1β (IL-1β) by macrophages coincides with the shift towards aerobic glycolysis, a metabolic process that mediates protection against drug-susceptible Mtb. Using a collection of MDR W-Mtb strains, we demonstrated that the overexpression of Mtb cell wall lipids, phthiocerol dimycocerosates, bypasses the interleukin 1 receptor, type I (IL-1R1) signaling pathway, instead driving the induction of interferon-β (IFN-β) to reprogram macrophage metabolism. Importantly, Mtb carrying a drug resistance-conferring single nucleotide polymorphism in rpoB (H445Y) can modulate host macrophage metabolic reprogramming (Howard et al. 2019). These findings transform our mechanistic understanding of how emerging MDR Mtb strains may acquire drug resistance single nucleotide polymorphisms, thereby altering Mtb surface lipid expression and modulating host macrophage metabolic reprogramming which needs to be thoroughly studied. This has implication in the efforts to successfully design new therapeutic targets and vaccines to prevent the spread of emerging MDR, as well as extensively and extremely drug resistant Mtb spread.